




For a drug to cross a membrane barrier it must normally be soluble in the lipid material of the membrane to get into membrane, also it has to be soluble in the aqueous phase as well to get out of the membrane. Most drugs have polar and non-polar characteristics or are weak acids or bases. For drugs which are weak acids or bases the pKa of the drug and the pH of the GI tract fluid and the pH of the blood stream will control the solubility of the drug and thereby the rate of absorption through the membranes, lining the GI tract.
Brodie et al. (in 1957) proposed the pH - partition theory to explain the influence of GI pH and drug pKa on the extent of drug transfer or drug absorption. Brodie reasoned that when a drug is ionized it will not be able to get through the lipid membrane, but only when it is non ionized and therefore has a higher lipid solubility.
Brodie tested this theory by perfusing the stomach or intestine of rats, in situ, and injected the drug intravenously. He varied the concentration of drug in the GI tract until there was no net transfer of drug across the lining of the GI tract. He then determined the ratio D:-
D = ![]()
Equation XII-1
i.e.
D = ![]()

Diagram XII-1 Showing Transfer Across Membrane
These values were determined experimentally, but we should be able to calculate a theoretical value if we assume that only non ionized drug crosses the membrane and that net transfer stops when [U]b = [U]g
The ratio [U]/[I] is a function of the pH of the solution and the pKa of the drug; as described by the Henderson - Hasselbach equation
For weak acids:-
pKa - pH = ![]()
and for weak bases:-
pKa - pH = ![]()
Brodie found an excellent correlation between the calculated D value and the experimentally determined values.
Even though the D values refer to an equilibrium state a large D value will mean that more drug will move from the GI tract to the blood side of the membrane. The larger the D value, the larger the effective concentration gradient, and thus the faster the expected transfer or absorption rate.
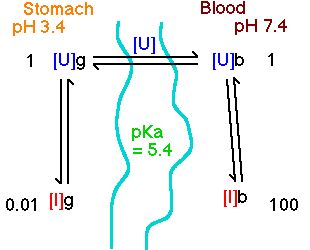
Diagram XII-2 Drug Distribution between Stomach and Blood
Compare D for a weak acid (pKa = 5.4) from the stomach (pH 3.4) or intestine (pH 6.4), with blood pH = 7.4
Stomach
[U]/[I] = 10pKa - pH = 105.4 - 3.4 = 102 = 100
i.e. [I] = 0.01 x [U]
Blood
[U]/[I] = 10pKa - pH = 105.4 - 7.4 = 10-2 = 0.01
i.e. [I] = 100 x [U]
Therefore the calculated D value would be
D = ![]()
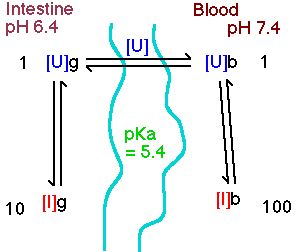
Diagram XII-3 Drug Distribution between Intestine and Blood
By comparison in the intestine, pH = 6.4
The calculated D value is (100+1)/(10+1) = 9.2
From this example we could expect significant absorption of weak acids from the stomach compared with from the intestine. Remember however that the surface area of the intestine is much larger than the stomach. This approach can be used to compare a series of similar compounds with different pKa values.
We have applied the pH - partition theory to drug absorption, later we will use this theory to describe drug re-absorption in the kidney.
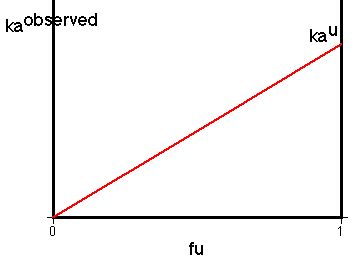
Figure XII-1, Plot of ka versus fu
With this theory it should be possible to predict that by changing the pH of the G-I tract that we would change the fraction non ionized and therefore the rate of absorption. This has some application in understanding drug absorption in overdose situations, but more readily used in relation to renal excretion.
Thus kaobserved = ku * fu assuming that the ionized species is not absorbed.
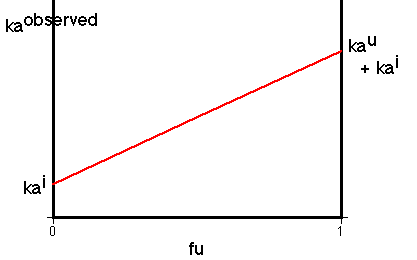
Figure XII-2, Plot of ka Versus fu for Sulfaethidole
For some drugs it has been found that the intercept is not zero in the above plot, suggesting that the ionic form is also absorbed. For example, results for sulfaethidole.
Maybe the ions are transported by a carrier which blocks the charge, a facilitated transport process.
Copyright 2001 David W.A. Bourne