
Figure 8.4.1 Linear Plot of Cp versus Time with ka = 3, 0.6, or 0.125 hr-1
return to the Course index
previous | next
Figure 8.4.1 Linear Plot of Cp versus Time with ka = 3, 0.6, or 0.125 hr-1
Click on the figure to view the interactive graph
Before going on to calculate the parameters ka, kel, and F from data provided we can look at the effect different values of F and ka have on the plasma concentration versus time curve. As ka changes from 3, 0.6 to 0.125 hr-1 the time of peak concentration changes to 1, 2.75 and 6.25 hour. Notice that with higher values of ka the peak plasma concentrations are higher and earlier.
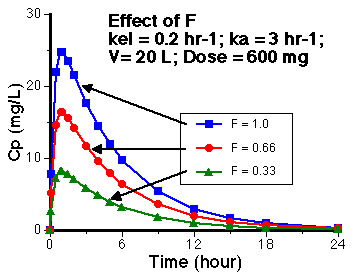
Figure 8.4.2 Linear Plot of Cp versus Time with F = 1, 0.66, or 0.33
Click on the figure to view the interactive graph
Changing F values is equivalent to changing the dose. Thus the higher the F value the higher the concentration values at each time point. Since the values of kel and ka are unchanged the time of peak plasma concentration is unchanged.
Thus, tpeak = 1, 1, and 1 hour. The same in each case.
Consider a drug with the parameter values in healthy subjects: F = 0.1; V = 766 L; kel = 0.105 hr-1; ka = 3.0 hr-1; CL = 74 L/hr. Plot a concentration versus time curve after an oral dose of 192 mg. Contrast this with the results obtained with the same dose given to a patient with severe cirrhosis. Use the parameter values: F = 1.0; V = 777 L; kel = 0.080 hr-1; ka = 3.0 hr-1; CL = 62 L/hr. Explore the problem as a Plot - Interactive graph. Pentikainen et al., 1978.
Consider a drug with the parameter values in healthy subjects: F = 0.30; V = 290 L; kel = 0.173 hr-1; ka = 3.0 hr-1; CL = 860 ml/min. Plot a concentration versus time curve after an oral dose of 80 mg. Contrast this with the results obtained with the same dose given to a patient with severe cirrhosis. Use the parameter values: F = 0.42; V = 380 L; kel = 0.063 hr-1; ka = 3.0 hr-1; CL = 580 ml/min. Explore the problem as a Plot - Interactive graph. Wood et al., 1978.
Consider a drug with the parameter values in original product: F = 0.30; V = 180 L; kel = 0.023 hr-1; ka = 5.0 hr-1; CL = 69 ml/min. Plot a concentration versus time curve after an oral dose of 500 mcg (0.5 mg). Contrast this with the results obtained with the same dose but with the new formulation. Use the parameter values: F = 0.75; V = 180 L; kel = 0.023 hr-1; ka = 5.0 hr-1; CL = 69 ml/min. Explore the problem as a Plot - Interactive graph. Danon et al., 1977.
A drug was administered as an IV injection, and oral solution and two tablet formulations (one rapid and the other a slow release tablet). A dissolution step was included to model the two tablet formulations. Parameters values included kel = 0.077 hr-1,V = 81.3 L, ka = 0.113 hr-1. The two tablet formulations required kd = 0405 hr-1 or 0.0261 hr-1. The dose were 25000 mg (Dose1 IV bolus), 20300 mg (Dose2 Oral solution), 19000 mg (Dose3 Oral rapid release tablet) and 21600 mg (Dose3 Oral slow release tablet). Simulate the concentration versus time curves after each dose. Bevill et al., 1977. Explore the problem as a Plot - Interactive graph.
return to the Course index
previous | next
Material on this website should be used for Educational or Self-Study Purposes Only
Copyright © 2001 - 2026 David W. A. Bourne (david@boomer.org)
 | Pharmacy Math Part Two A selection of Pharmacy Math Problems |
 |